|
پروفسور محمد حسین سلطان زاده
استاد
دانشگاه علوم پزشکی شهید بهشتی
متخصص کودکان ونوزادان
طی دوره بالینی عفونی از میوکلینیک آمریکا
دبیر برگزاری کنفرانس های ماهیانه گروه اطفال
دانشگاه علوم پزشکی شهید بهشتی
|
اقای دکتر
شاداب صالح پور
فوق تخصص غدد ومتابولیک اطفال
به اتفاق اعضای
هیئت علمی
بیمارستان
لقمان حکیم
|

اقای دکتر
شاداب صالح پور
فوق تخصص غدد ومتابولیک اطفال
Answer
Differential Diagnosis
This 6 year old boy had profound motor and intellectual deterioration, with
cortical and pseudobulbar manifestations, beginning at 1-2 years of age and
subsequent onset of a mixed-seizure disorder (possibly myoclonus epilepsy). In
addition, he was said to have manifested discrete, presumably foveal, patches of
cherry-red retina in both retinas. These deficits are referable bilaterally to
the retina, cerebral cortex, supranuclear descending fiber tracts (especially at
the level of the midbrain), and cerebellum or its connections. It is important
that no marked dysmorphism, organomegaly, or dysostosis was found.
This boy's early intellectual development was not entirely normal. He had a
relatively slow phase of psychomotor decline between the ages of two and five
years, with frequent falls. The history does not permit us to differentiate
among ataxia, dystonia, weakness, spasticity, spells of atony, and visual
difficulties as the cause of the falls. Repeated bouts of tonsillitis permit us
to rule out ataxia telangiectasia.
Shortly before the patient's fifth birthday, seizures developed. They were
initially tonic and subsequently atonic, and they increased in frequency. In a
child with ataxia and neurologic degeneration, this sequence suggests a
progressive myoclonus epilepsy. Poor feeding (despite a good appetite and
preserved lower-cranial-nerve function), barely intelligible speech, uncertain
proximal strength, intact sensation of pain, pyramidal signs, and inability to
maintain a vertical posture are nonspecific findings in childhood
neurodegenerative diseases. We have three important diagnostic clues, all of
which are related to the visual system: the presence of cherry-red spots,
limitation of the gaze, and reduced visual acuity, with concentric reduction of
the visual field.
A few test results are more helpful for ruling out diagnostic possibilities than
for supporting their inclusion in the differential diagnosis. In this case, it
is important that the cerebrospinal fluid protein level and immune profile and
the peripheral-nerve conduction velocities were normal. On
electroencephalography, the voltage of somatosensory evoked potentials was very
high; this finding is most helpful in suggesting a diagnosis.
The axial T2-weighted MRI studies show two abnormalities. The white
matter is hyperintense on T2-weighted images — a finding that is
nonspecific but very abnormal in a person of this age. The ventricles and the
sulci are prominent, indicating the presence of generalized atrophy. On the
coronal T1-weighted images, there is a thin, periventricular band of
hypointensity bilaterally; it is similar in intensity to the cortical gray
matter. The distinctive visual abnormalities in this case are specific
diagnostic clues, but they lead in different directions.
The term “cherry-red spot” refers to the normal hue of the fovea, which may be
thrown into startling relief in some storage disorders that produce a pallid
perifoveal ring. It is a characteristic finding of lysosomal storage diseases.
The hue of the normal fovea, and likewise that of the cherry-red spot, varies
according to the pigmentary-gene complement and ranges from pale pink to the
cherry black that may be seen in dark-skinned persons. If an abnormal perifoveal
ring is present, we can be almost certain that the patient has 1 of about 15
storage diseases. Cherry-red spots may result from Fabry's disease or from
vascular disorders such as stroke, incontinentia pigmenti, trauma, or toxicity
(in which the spots are often unilateral or asymmetric). All these conditions
can be ruled out in this case. Other macular abnormalities — particularly the
bull's-eye maculopathy associated with late-onset infantile neuronal ceroid
lipofuscinosis — may resemble cherry-red spots, so a precise description of the
lesion is essential.
A finite group of diseases in which cherry-red spots are found include the
sialidoses, GM1 and GM2 gangliosidoses, Niemann–Pick
disease (Crocker groups A through D but not E and F), Farber's
lipogranulomatosis, and metachromatic leukodystrophy. Cherry-red spots are not
present in all cases of these disorders, and in cases in which they are present
the spots may appear, disappear, or change in hue. Their appearance and
disappearance are influenced by the accumulation of storage material in the
lysosomes of the perifoveal ganglion cells. Changes in hue may be related to
secondary macular deterioration, as in some patients with Tay–Sachs disease.
The appearance of the perifoveal change is important in the differential
diagnosis and depends on the types and amounts of material stored in the
perifoveal ganglion cells. In Tay–Sachs disease and Sandhoff's disease, the
considerable amount of stored material produces an opalescent white ring. In
Niemann–Pick disease, the ring is more diffuse and may have less distinct
margins; it may even be patchy. The faint, gray ring of Farber's
lipogranulomatosis and of metachromatic leukodystrophy may render the cherry-red
spot more difficult to visualize.
We can rule out many of the aforementioned diseases, either because they are
manifested earlier or later than the disease in this case or because the pattern
of neurologic dysfunction is dissimilar to that observed in this patient. Other
diseases that can be ruled out are associated with storage of gangliosides or
other substances in other parts of the body, producing a so-called
mucopolysaccharidosis phenotype that includes facial dysmorphism, organomegaly,
dysostosis multiplex, and inguinal hernias.
Patients with group B Niemann–Pick disease have massive organomegaly and are
neurologically normal. The few cases of group B disease involving mild ataxia or
mental retardation have proved to be misdiagnosed cases of group C disease. Type
I sialidosis, myoclonus, cherry-red spots, and severe impairment of motor-system
function develop in adolescence, and the vision and intellect typically are
normal. Patients with type II sialidosis (congenital or infantile sialidosis or
galactosialidosis) have an extreme mucopolysaccharidosis phenotype. Type I GM1
gangliosidosis (previously called Norman–Landing disease) is characterized by
severe neurologic failure in infancy with a marked mucopolysaccharidosis
phenotype. Both acute infantile forms of GM2 gangliosidosis (classic
Tay–Sachs disease and Sandhoff's disease), the most common causes of cherry-red
spots, must be ruled out in this case, regardless of the presence or absence of
organomegaly. They have an infantile onset with macrocephaly, and the type and
severity of their neurologic manifestations differ from those in the present
case.
None of the six variant forms of Farber's lipogranulomatosis (including type IV,
which may be associated with cherry-red spots) belong in the differential
diagnosis. Metachromatic leukodystrophy may also be associated with cherry-red
spots and with ataxia, speech deterioration, and episodes of hypertonus, which
could have caused this patient to lose his balance and fall. The absence of any
peripheral-nerve involvement rules out this diagnosis.
Subacute GM2 gangliosidosis of either the Tay–Sachs or Sandhoff's
biochemical profile has a late infantile or juvenile onset and is associated
with ataxia, incoordination, loss of speech, loss of self-care skills, dementia,
spasticity, and seizures that progress at variable rates. Cherry-red spots are
seldom present, however, although optic atrophy and retinitis pigmentosa may
develop. Although most patients with group C Niemann–Pick disease have
organomegaly, it may be overlooked. The characteristically mild delay in early
development and the frequent falls, speech difficulties, ataxia, and dysphagia
tend to occur at the same ages as they occurred in the child in the current
case; however, children with group C Niemann–Pick disease usually have neonatal
jaundice.
This patient's gaze difficulties are an important reason to retain subacute GM2
gangliosidosis and especially Crocker group C Niemann–Pick disease in the
differential diagnosis. The features of this case are consistent with those of
Wernicke's pseudo-ophthalmoplegia, a manifestation of pseudobulbar palsy.
Pseudobulbar palsy is also the likely explanation of this child's speech and
feeding difficulties. Vertical supranuclear limitation can be localized to the
midbrain pretectal portion of the rostral medial longitudinal fasciculus. Many
of the known causes of this dysfunction can be dismissed on the basis of the
history in this case.
Supranuclear gaze palsy, particularly that affecting the vertical gaze (downward
saccades more than upward), is particularly characteristic of Crocker group C
Niemann–Pick disease. Most cases of so-called juvenile dystonic lipidosis, which
also characteristically produces supranuclear vertical-gaze palsy, have also
been examples of group C Niemann–Pick disease, although some are atypical cases
of neuronal ceroid lipofuscinosis. There is a single case report of a
supranuclear gaze palsy in a patient with Kufs' disease, or adult-onset neuronal
ceroid lipofuscinosis. Supranuclear gaze palsy has also been described in
late-onset, chronic variants of GM2 gangliosidosis. The absence of
organomegaly weighs against group C Niemann–Pick disease or juvenile dystonic
lipidosis.
Pseudobulbar gaze palsy may occur in metachromatic leukodystrophy, but usually
at a late stage of severe illness and in association with bulbar signs. In the
current case, the normal level of protein in the cerebrospinal fluid and the
normal nerve-conduction velocities argue against the infantile form of
leukodystrophy. A case of infantile Krabbe's disease with a cherry-red spot has
been described.
Diminished visual acuity with restriction of the visual fields suggests the
presence of tapetoretinal degeneration and would be an unusual manifestation of
a disease producing cherry-red spots. Visual loss is characteristic of
gangliosidosis — hence the former labeling of Tay–Sachs disease as “amaurotic
idiocy” — but it is predominantly cortical rather than retinal. Retinal changes
may be seen in late-onset GM2 gangliosidosis, which may in some
instances resemble those of neuronal ceroid lipofuscinosis. Retinal degeneration
in mitochondrial encephalomyopathic disease is not suggested by the features of
this case, and mitochondrial degeneration produces ophthalmoplegia rather than
pseudo-ophthalmoplegia.
Neuronal ceroid lipofuscinosis accounts for the remaining disorders formerly
classified as amaurotic idiocy. These conditions are so difficult to distinguish
from the gangliosidoses that we must rely on the presence or absence of
cherry-red spots to diagnose the infantile forms and on electroretinographic
findings to diagnose forms with a later onset. In the current patient, the
retinal disease that gave rise to the visual-field loss as well as the
progressive myoclonus epilepsy are far more consistent with the late-onset
infantile (Janský–Bielschowsky) form of neuronal ceroid lipofuscinosis than with
any of the diagnoses associated with cherry-red spots. This diagnosis also
provides an explanation for the large evoked potentials and the high-amplitude
electroencephalographic changes in this case, which could otherwise be explained
only by type I sialidosis.
Thus, an important question in this case is whether the patient's cherry-red
spots are actually a ganglion-cell storage disorder producing bull's-eye
maculopathy of the type associated with late-onset infantile neuronal ceroid
lipofuscinosis. The late-onset infantile form of neuronal ceroid lipofuscinosis
causes macular dystrophy, rather than the retinitis pigmentosa found in patients
with later-onset ceroid lipofuscinosis (Batten disease or Vogt–Spielmeyer
disease). The bull's-eye macula is often clearly demarcated from a surrounding
area of hypopigmented retina and usually shows pigmentary clumping, particularly
at its margins. This child may have had relatively dark skin, which may be
associated with unusually uniform pigmentary clumping at the macula, resulting
in a usually convincing, false cherry-red spot. First described in 1933, the
bull's-eye maculae of Janský–Bielschowsky disease may be brown, reddish brown,
or mottled and are sometimes surrounded by a gray zone.3
Photographs of the retina were taken only after the diagnostic procedure had
been performed.
The photographs show a gray ring, or halo, in the perifoveal area, setting apart
a fovea that appears cherry red.
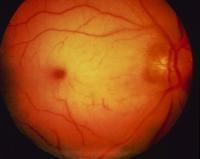
However, the pigmentation of the fovea is not uniform, suggesting pigmentary
clumping at the margins. These features indicate that the lesion is better
interpreted as bull's-eye maculopathy, which is typical of late-infantile
neuronal ceroid lipofuscinosis. This interpretation is more consistent with this
patient's epilepsy, visual-field restriction, and electrophysiological
abnormalities than with disorders associated with true cherry-red spots. If we
return to the fact that this child had high-amplitude visual evoked cortical
responses, the diagnosis is all but certain.
The patient's ethnicity (Iranian- Middle Eastern origin) also suggests this
diagnosis. In some areas of the Middle East, the rate of first-cousin marriages
exceeds 50 percent, and autosomal recessive ceroid lipofuscinosis is among the
most common neurodegenerative diseases.
The most direct method for establishing this diagnosis is electroretinography.
If there is no retinal response to electroretinography, electron-microscopical
examination of biopsy specimens of the conjunctiva or of eccrine-gland–containing
tissues may show the diagnostic curvilinear bodies containing abnormal lysosomal-storage
material. Genetic testing could also confirm the diagnosis.
When the patient was examined first, visualization of the fundi was not
completely achieved, except to note that they appeared pale. The discovery of
cherry-red spots by an ophthalmologic consultant turned our attention to the
neuronal ceroid lipofuscinosis. We favored the diagnoses of Sandhoff's variant
of GM2 gangliosidosis and neuronal ceroid lipofuscinosis. However,
another possibility, mitochondrial encephalomyelopathy, dictated our choice of
diagnostic procedure.
Clinical Diagnosis
Neuronal ceroid lipofuscinosis, late-onset infantile subtype (Janský–Bielschowsky
disease).
Pathological Discussion
The diagnostic procedure was a biopsy of the left quadriceps muscle and of the
sural nerve. Microscopical examination of the muscle-biopsy specimen revealed
only slight variation in the size of the myofibers.
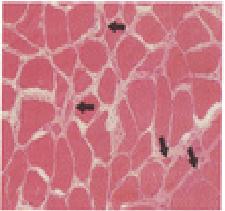

An acid phosphatase stain showed reddish granular deposits within the myofibers,
indicating an increase in lysosomal activity. Light-microscopical examination of
the nerve was unremarkable.
Neuronal ceroid lipofuscinosis is a heterogeneous group of progressive,
neurodegenerative disorders that occur in children and, less commonly, in
adults. In most cases, these disorders have an autosomal recessive mode of
inheritance, with rare cases of autosomal dominant inheritance occurring in
adults. The overall incidence of neuronal ceroid lipofuscinosis worldwide is 1
in 12,500 live births. Clinically, these disorders are characterized by visual
loss, epilepsy, and psychomotor deterioration. The classification of these
disorders is based on age at onset, clinical features, morphologic features on
ultrastructural examination, and genetic subtype.
The characteristic morphologic lesions of neuronal ceroid lipofuscinosis are the
loss of neurons and widespread intracellular accumulation of lipid pigment
within neurons and other types of cells, including lymphocytes and cells of the
vascular endothelium, sweat-gland epithelium, and muscle. The degree of neuronal
loss varies according to the subtype of the disorder. Despite the accumulation
of intracellular lipid pigment, neurons do not have the “ballooned” appearance
that is seen in other storage disorders. The lipid pigment characteristically
stains with Luxol fast blue, Sudan black, and periodic acid–Schiff. In
ultraviolet light, the material has a yellow to silvery autofluorescence that
differs from the orange autofluorescence of lipofuscin. Although the
ultrastructural characteristics of the lipid pigment are not pathognomonic of
specific subtypes of the disease, particular forms are present in each subtype.
Curvilinear bodies are the only form in which lipid pigment is found in classic
late-onset infantile neuronal ceroid lipofuscinosis (the CLN2 subtype). The
storage material is membrane-bound, a feature that is consistent with its
localization within lysosomes. Subunit c of mitochondrial ATP synthase is the
primary component of the lipid pigment in the CLN2, CLN3, and CLN4 subtypes,
whereas sphingolipid activator proteins A and D predominate in the CLN1 subtype.
In the current case, the boy's age at the onset of disease, the clinical
presentation, and the ultrastructural features are most consistent with the
classic late-onset infantile subtype of neuronal ceroid lipofuscinosis (the CLN2
subtype). The CLN2 gene is located on chromosome 11p15. In one study, an
intronic mutation (in which cytosine was substituted for guanine at position
T523-1), affecting a splicing junction, and a nonsense mutation (in which
thymine was substituted for cytosine at position 636) were found, singly or
together, in 69 percent of patients. The CLN2 gene product is homologous
to tripeptidyl peptidase I (TPP-I), a lysosomal, pepstatin-insensitive
exopeptidase that cleaves tripeptides from the N terminal of oligopeptides and
proteins. TPP-I activity is deficient in the fibroblasts and brain tissue of
patients with this type of the disorder. It has been hypothesized that subunit c
of mitochondrial ATP synthase is a substrate for TPP-I and that the accumulation
of subunit c is a result of deficient TPP-I activity.
Because the abnormal lipid pigment accumulates in many types of cells,
ultrastructural examination of many tissues may be diagnostically useful.
Prenatal diagnosis is based on the examination of chorionic or amniotic cells
for the presence of the abnormal storage material or decreased enzyme activity
or by mutational or haplotype analysis and comparison of the results with those
in the reference patient
Tests for arylsulfatase A, very-long-chain fatty acids, and β-galactosidase were
negative. Oligosaccharide screening was negative. Genetic tests for two of the
mutations implicated in neuronal ceroid lipofuscinosis were negative.
Electroretinography demonstrated attenuated responses to stimulation that were
consistent with advanced diffuse retinal degenerations preferentially affecting
cone photoreceptor function, strongly favoring a diagnosis of neuronal ceroid
lipofuscinosis.
The patient had a younger sister, who was about seven months old at the time of
his diagnosis. The sister appeared clinically normal at that time, but we have
no further information.
The FINAL Diagnosis
تشخیص
Neuronal ceroid lipofuscinosis,
late-onset infantile subtype
معرفی کیس توسط
اقای دکتر دکتر
زندی
رزیدنت بخش اطفال بیمارستان لقمان

خانم دکتر بیگلری
رزیدنت بیمارستان مفید

اقای دکتر سنجری
رزیدنت بخش اطفال بیمارستان امام حسین

خانم دکتر بیگلری
رزیدنت بیمارستان مفید
IN THE NAME OF GOD
Problem list :
�a
6y/o boy with seizure disorder& progressiv neuro developmental delay
�NL
development up to 1.5 y/o
�
Seizure responsible to anti convulsant drug(Na Val)
�Limitation
of the elevation of eyelids(supra nuclear vertical gaze palsy)&impaired
saccade movment. Dysarthria- dysphagia-FTT- ataxic gait-
�Decreased
visual acuity& bilateral cherry red spot
�DTR
+++ ; Babinski sign +; without pripheral neuropathy
�EEG
not significant ; CSF NL; VEP nl; SSEP ab.nl
�MRI:
priventricular hyperdense banb like sign & diffuse cerebral and cerebellar
atrophy
Cherry red spot DDX:
�GM1
�GM2(
tay sachs – sandhoff)
�Nieman
pick(A ‘ B)
�Nieman
pick (C ‘ D)
�Gauscher
disease
�Methachromatic
leukodystrophy
�Krabbe
disease
�Ceroid
lipofocinosis
Leber
congenital amaurosis
�Farber
disease
�Sialidosis(
1 ‘2 )
�Galactosialidosis
�MPS
1(Hurler dis)
�MPS
7 (sly disease)
�Hallervorden
spatz syn
�Wolman
disease
�Retinal
artry occlusion
�Quinin-
Dapson
Vertical supranuclear gaze palsy :
�Midbrain
strok
�Pineal&
metastatic tomor
�Inflamation
&infection disorders
�Demyelinating
disorders(MS)
�Wilson
disease
�Kericterus
�Wernicke
syn
Metabulic
disease:•Bassen
–kornzweig syn(abete lipoproteinemia)
•Niemann-
pick C disease
•Tay
sachs disease
•Gauscher
disease
•MSUD
disease
•Hyperglycinuria
Symptomatic(secondry) myoclonus(not defined by occurrence of seizure)
Progressive
myoclonus epilepsy: Baltic myoclonus
Spinocerebellar
degeneration: Ramsay-Hunt & Friedreich &ataxia telangectasis
Basal
gangelia degeneration
Dementias&infection&
toxin& malabsorbtion& metabolic dis(MCD-BD- electrolite dis&…)
Focal
nervous sys damag
Storage
dis:
Lafora body dis
GM2
gangliosidosis(late inf & juvenile)
Tay sachs
Gauscher
dis(non inf- neuropathic form)
Krabbe dis
Neuronal ceroid
lipofocinosis Sialidosis(1.2
abetalipoproteinemia
•Malabsobtion(fat)-
steatorrehea-neurologic manifestation-retinitis pigmentosa –s.n
palsy
•Ataxia
– pripheral neuropathy-( sensory motor neuropathy [2-6y]
•cherry
red spot & no malabsobtion
MSUD disease:(classic)
•First
48h of birth
•
with PF- irritability-vomiting
•Ketonuria-keto
acidosis& lethargy
•4days
•Seizure-dystonia&
apnea
•wilson
•kernicterus
•Hyper
glycinuria
•Hallervorden
spatz
•Movment
disorders
•Wernicke
syndrom
•B1&B12
deficiency-megaloblastic anemia
•Apathy-
Dementia Memory impairment
•MPS
1-7
•Course
face
•Organomegaly
&cardiac invulvment
•Bone
invulvment
•
eye invulvment&hydrocephaly
�hallervorden
spatz syn: Since 3-4 y dystonia &
dysphagia –dysarthria- ataxia- seizure disorder-rigidity- dementia- eye of the
tiger in MRI . Cherry red spot + -with
out VSN gaze palsy
•Farber
dis: articular manifestation- organomegaly- cardiac-
horsness – CR spot+ -
with out VSN gaze palsy
�Krabbe
disease:( globoid cell dystrophia)
Infantile:<6mo ‘ microcephaly’ optic atrophia’ irritibility’ generalized
seizure’ death before 2y
Juvenile: >2y – nl
inteligence up to 3y- gradually regressed- optic atrophia-CR spot + priphral
neuropathy- psychiatric disordes-
without SNV gaze palsy
•Leber
congenital amaurosis:Retinitis pigmentosa-
nephronophtiasis-CR spot+
without SNV gaze palsy
Metachromatic leukodystrophy:
�
late infantile:the most- 12-18 mo- irritibility- unable
to walking- jenorecuartom- hyper extention of knee-
quadriplegia- hypotonia- optic atrophia- myoclonic S-
death up to 10y
�Juvenile:20y-
this process slowly- psyciatric disorders –psychosis
�Adult:
20-30y- psychosis- dementia- epilepsy – optic atrophia-
(
increase
CSF Pr)
Cherry red spot+
supra nuclear gaze palsy _
Sialidosis:
�Type
1: 2th decade with decrease visual acuity - myoclonus
induced with any stimulant & not response to anti
convulsunt drugs - cludy cornae- -CHERRY
RED spot ++
SNVG palsy _
�Type
2: coarse faces- disostosis moltiplex in 1th y- mild to
mod MR – HSMG- cardiac-
CHERRY RED spot ++
SNVG palsy _
Galactosialidosis: HSMG- coarse face- disostosis
moltiplex -
GM1:
�TYPE
1: early infantile- HSMG-edema- angiocratoderma-
psychomotor retardation& TC seizure(up to 6 mo)- ab.nl
faces- bone involvment such as MPS- death 3-4y
�
Type 2: childhood- neurologic manifestation( ataxia-
dysarthria- MR – spasticity)- degree of bone &visceral
low
CHERRY RED spot ++
SNVG palsy _
Sandhoff dis(GM2):
�HSMG-cardiac
and bone involvment low- macrocephalia-doll’s face-
seizure- juvenile form: ataxia- dysarthria &
psychiatric disorder
SNVG palsy (_ )Cherry
red spot suspected
Late
infantile onset: 6mo-2y. Regression of motor skill- gait
problems- ataxia- hypertonia- babinski+- optic atrophia-
death in 5-6y.
Juvenile:
4-8y- gait problems- ataxia- priphral neuropathy-
psychiatric disorders- death after 6y
Tay sachs(GM2):
�Infantile:
NL up to 4-5 mo NL- low eye contact- hyper acausis-
macrocephalus without hydrocephalus- sever neurological
degeneration- progressive ataxia- dysarthria up to 4-5
y- organ involvement (low or neg)
SNVG palsy+ cherry red spot +
�Juvenile:
childhood- spasticity- clumsiness- ataxia- seizure-
progressive decrease visual acuity-cherry
red spot(+) SNVG palsy+
psychiatric disorder
Ceroid lipofocinosis:
�Infantile
type:>1y – NDD- myoclonic seizure- ataxia- blindness-
optic atrophy- death in 10y
�Late
infantile type: common type- 2-4y - microcephaly-
myoclonic seizure-decrease of visual acuity- ataxia-
dementia-pripheral black pigment in retina(spicle bone)-
drop attack such as this patient
but
without SNV gaze palsy
�Juvenile
type:5-10y- progressive visual acuity-
Without
SN palsy
The most
common neurodegenerative dis in children
Gauscher disease:
�The
most common lysosomal storage disease.
Type
1:
CNS
intact-
90%
of GD patients have non neuropathic form– neurological
involv(--) but
visceral
involv(+) ( bone’ SMG’ anemia’ trombocytopenia ’
osteopenia’ liver fibrosis’ pul HTN ) - 12-24mo often
childhood- slowly progression-
Type2:
acute neurological
involv (+)
– low
visceral involv - bone -
congenital ictiosis-
sever MR- generalized seizure- death <2y
Gauscher disease:
Type
3: neurological involv+
a:
childhood -visceral - myoclonus-apathia- dementia-
anemia- osteopenia – death 20-30 y
b: cildhood-
sever
hematologic dis-
sever
osteopenia-
supra nuclear gaze palsy
-life
time : low
c:
childhood- sub acute- hematologic - bone -
supra nuclear gaze palsy-
cardiac involv+ - life time: low
Niemann pick disease:
�Type
A: 1 mo- HSMG- ILD-feeding problem- motor skill -
priphral neuropathy- progressive neurological dis- LDL&TG
- death 2-3y-cherry
red spot(+)
but SNVG palsy
�Type
B: infancy or childhood- HSMG- short stature- delay
skeletal maturation- cerebellar sign- nystagmus- extra
pyramidal sign- MR- death up to adulscent -
Niemann pick disease:
�Type
C: onset varriable (uterin- infantile- childhood- adult)
Infantile:
prolonged icter- liver dis- hypotonia- respiratory
distress
Childhood:
most patients have disease onset in middle to late
childhood after nl early development. Cerebellar & gait
problem-
slowly progressive cognitive detoriation-
dystonia- dysarthria- dysphagia- seizure-
SNVG
palsy-
death
20-30y
Adult
: psychiatric( dementia- depression- bipolar-
schizophrenia)
DDX of CR spot & SN palsy:
•Niemann
pick C
•Tay
sachs (juvenile)
•Gauscher
disease 3C
Diagnoses of this case:
Nieman
pick C
Tay
sachs disease(juvenile)
Neuronal
ceroid lipofocinosis
Gauscher
disease 3C

اقای دکتر سنجری
رزیدنت بخش
اطفال بیمارستان امام حسین


















