|
پروفسور محمد حسین سلطان زاده
استاد
دانشگاه علوم پزشکی شهید بهشتی
متخصص کودکان ونوزادان
طی دوره بالینی عفونی از میوکلینیک آمریکا
دبیر برگزاری کنفرانس های ماهیانه گروه اطفال
دانشگاه علوم پزشکی شهید بهشتی
|
خانم دکتر مژگان هاشمیه
فوق تخصص خون انکولوژی
خانم دکتر آزاده کیومرثی
رزیدنت
اطفال بیمارستان مفید
آقای
دکتر امیرحسین حسینی
رزیدنت
اطفال بیمارستان شهدا
|
خانم دکتر مژگان هاشمیه
Hemophagocytic lymphohistocytosis (HLH)
is
a potentially life threatening condition occurring as a:
familial
disease (FHLH) or
secondary
to marked immunological activation during viral, bacterial and parasitic
infections, malignancies, rheumatologic conditions or immune
deficiencies with cytotoxic T and/or NK-cell dysfunction .
Although FHLH is an autosomal recessive disease that
affect immune regulation, sporadic cases with no obvious family
inheritance occur.
All organ systems might be affected in HLH.
Eventually multiple organ dysfunction syndrome (MODS)
develops and death occurs.
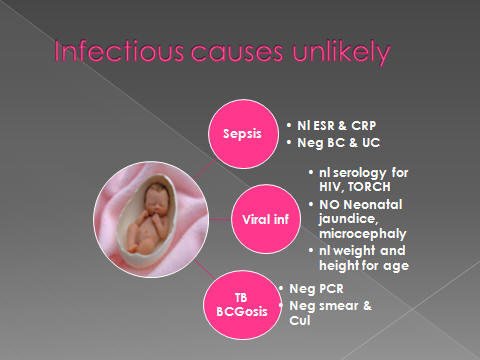
Clinical presentation of HLH might be confused with :
sepsis
metabolic disorders,
immune deficiency syndrome
&BCGeosis.
The diagnosis of HLH can be established if 1of either
2 below is fulfilled
A) A molecular diagnosis consistent with HLH
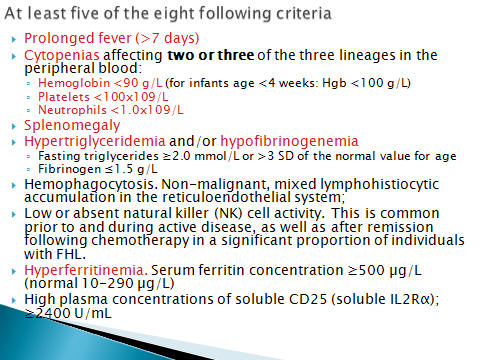
B) Diagnostic criteria for HLH fulfilled five of
eight criteria below
1- Splenomegaly
2- Cytopenia (affecting >2 of 3lineages in the
peripheral blood)
3- Hypertriglyceridemia and / or hypofibrinogenemia:
fasting TG >265 mg/dl fibrinogen <1.5 gr/dl
4- Hemophagocytosis in bone marrow or spleen or lymph
nodes with out evidence of malignancy
5- Low or absent natural killer cell activity
6- Ferritin > 500 µg /l
7- Soluble CD25 (IL2 receptor) > 2400/ml
8-fever
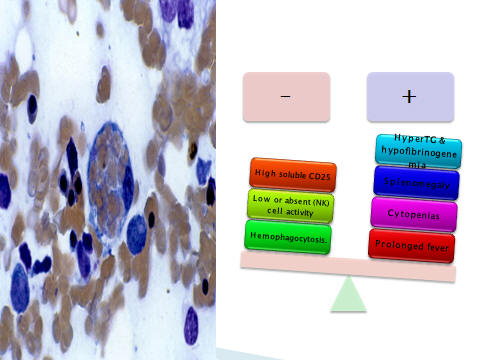
Five of the eight criteria for FHLH in our patient
1-Fever
2-Hepatosplenomegaly
3- Bicytopenia with a total white count at 6.1× 103
/ µl , and 14% PMN (absolute neutrophil count :714),
Hb: 8.7 gr/dl and platelet count:55×103/µl
4-Ferritin: 1135 µg /l (normal range: 25-200 µg /l)
5- TG: 794 mg/dl (normal range <110 mg/dl),
fibrinogen was less than 2 g/dl (normal range: 2.5-4 g/dl).
Considering the positive familial history of death in her sibling with
similar presentation and familial relationship of parents , FHLH was the
first suspected diagnosis1,
Some
metabolic diseases like lysinuric protein intolerance
1,9
have a
similar clinical presentation with HLH .
Therefore we checked our patient metabolic profile including serum levels of
lactate, ammonia, pyruvate also chromatography of amino acid and sugar in
blood and urine.
All the
results were normal so metabolic disorder was ruled out .
Because of worsening of clinical condition in our patient and her sibling
after BCG injection, another differential diagnosis was immune deficiency
syndromes .
we
performed bone marrow flow cytometry immunophenotype analysis. The results
were not specific and did not support this diagnosis.
Considering some reports of association between tuberculosis and final
diagnosis of secondary HLH , PCR and culture of CSF, liver biopsy, BM
aspiration and ascitis fluid were performed.
No
evidences of TB were found.
The
clinical findings in children with infection associated HLH are similar to
those in FHLH .
The
most common agent causing this syndrome is viruses, predominantly the herpes
group viruses including EBV, HSV, and CMV .
A
search for these etiologic agents were performed in our patient .She was
found to be negative for EBV, HIV, CMV and rubella virus.
A
distinctive diagnosis of FHLH can be made if there are genetic defects
involving the
perforin
gene on chromosome 9q21.3 locus (FHLH type 1 ),10q
21-22mutations (FHLH type 2).
Perforin acts by perforatings the cytolytic target cell membrane in turn
initiate the apoptotic cell death pathway
1,9.
The
other genetic defects are inactivating the
MUNC 13-4
gene which is essential for cytolytic granule fusion at
chromosome 17q 25 (FHLH type 3) and mutations in
syntaxin
11
gene which is located on chromosome 6q24.
Hemophagocytosis might be found in the first bone marrow aspiration of a
FHLH patient but the absence of it will not rule out the diagnosis.
In
previous literature some cases of FHLH without hemophagocytosis has been
reported.
In our
case, the bone marrow aspiration was performed two times and liver biopsy
one time that did not revealed any site of hemophagocytosis.
خانم دکتر
آزاده کیومرثی





آقای
دکتر امیرحسین حسینی
A 52
day old girl with high grade fever & poor feeding who expired in hospital on 2nd
week of admission
History
lHx.
l52
d/o girl with fever & poor feeding
since 1week prior to admission.
lPt.
was on Ampicillin, ceftriaxone, gentamicin 1week prior to addmission
lReffered
due to Bicytopenia
l
lPMH.
l3rd
sibling , GA= 38wk, elective C/S ,G3P3L2A0D1,
lB.wt=3500g,
HC=35, Lt=50 cm
lNICU
admission for 7 days due to pneumnia
lVaccination
history is complete
lFamily
hx.
Parents are relatives
l1st
sibling is 9 y/o girl and in good health condition
l2nd
sibling expired on 2mo of age due to fever, splenomegaly &pancytopenia
(after vaccination)
Physical examination
lOn
admission; ill, pale & irritable
lv/s:
T=38 c /RR=48/ PR= 120bpm/BP=90/50 mmHg/ O2 Sat. without O2=88-98%
l
wt= 5400g, HC=38cm, Lt.=53cm
lAnt.
Fontanel= 1.5*1.5 cm----
l
Post. Fontanel= tip of finger
lA
firm LN (1*1cm), mobile, non tender, in Rt. axilla
lLung:
clear (bilat.) with mild IC retraction
lHeart:
nl
lAbdomen:
distended, spleen can be palpated in umbilical area, liver span=8cm.
lMuscle
tone is nl
lGood
eye contact
lOther
examinations are nl
Hospital course
lPt.
treated with
vancomycin +meropenem
l4
days later pt’s respiratory distress & abdominal distension exacerbated
lAbdominal
sonography:
hepatomegaly (98mm),
splenomegaly (99mm),free fluid in abdomen, Pleural effusion in Rt. hemithorax
without LAP.
Lab. data
lWBC=6100/micl
(P=14%, L=83%), Hb=8.7gr/dl, Hct=25.6%, Plt=55000/micl, MCV=77.2fl , RBC=
3660000/micl,
Retic= 1%
lESR=
12 CRP=+1
lAST=108
u/l ALT=70u/l LDH=950IU/l, Alb=3.1g/dl
lBUN=10mg/dl,
Cr=0.5mg/dl, UA=6mg/dl, BS=70gr/dl,
lPT=12s,
PTT=47s
l
lTG=
240 mg/dl
lFibrinogen
level: nl
lFerritin
:nl
lABG:
PH=7.46, Pco2=33.6, Po2=58.3, HCO3=24.2
lNa=137meq/l,
K=4.1meq/l,
lU/A:
nl U/C :neg.
lB/C:
neg S/E: nl
lAcitic
fluid:
glu.=67mg/dl,
pro.=3.6g/dl, WBC=250 (P=20%,MN=80%)
RBC=5200, LDH=643
lCXR:
bilat. Perihilar haziness
lPBS:hypochromia,
anisocytosis, leukopenia, partial lymphocytosis, thrombocytosis
lBMA
(2 times with 10 days interval):
60% cellularity with
maturation arrest at myeloid series without evidence of hemophagocytosis. No
blast, no erythrophagocyte
lBM
immuno-phenotyping:
60% lymphoid
immune-population of total cells, half of them are mature T-cells with
reverse CD4/CD8 ratio,
B-precursors expressing CD19, HLA-DR & variable expression of CD20
lMetabolic
profile
(Before pc transfusion): including lactate, ammonia, pyrovate
& chromatography of A.A. & sugar in blood & urine were Nl
lToRCH
study: nl
lCMV,
HSV, EBV, HCV, HIV : neg
lPCR,
smear & culture of ascitic fluid, CSF & BM were neg. about
acid fast bacilli
l
lLiver
Bx: chronic active hepatitis
l
lBone
survey: Nl
l
Lab. data on
10th
day of admission
lTG=794
mg/dl (nl<110)
lFerritin
=1135 micgr/dl (nl. 25-200)
lFibrinogen
level: <2 gr/dl (nl.2.5-4)
l
On 14th
day
lCardiopulmonary
arrest
lCPCR
was not successful
lNo
permission for autopsy
l
lLast
lab. Data:
CBC: WBC=6100
(P=18%,L=80%) plt=33000, Hb.=9.5
ABG: PH=7.57,
pco2=32.4, Hco3=29.7 Sat. O2=87.7%
l
Problem list
A 52 d/o girl, term,
with fever, poor feeding (since 7 days prior to admission), irritability,
abdominal distension, who had the
following
data,problems, and
Para clinic points:
l
Fever + poor feeding
lparents
are relatives
lPositive
family history of a similar death in previous sibling
lNo
skin lesion, no congenital deformity
lHepatosplenomegaly
+ ascetic fluid
lBicytopenia
lHypertrigeliceridemia
lhypofibrinogenemia
l
high ferritin level
lBM
immuno-phenotyping:
Reverse CD4/CD8 ratio
lMetabolic
profile : nl
lABG:
nl
lNl.
Blood Sugar /NL. biochemistry
lNl.
U/A,
l
mildly elevated liver enzymes
lToRCH
study: nl
lCMV,
HSV, EBV, HCV, HIV : neg.
lBMA:
without evidence of hemophagocytosis. No blast, no erythrophagocyte
lPCR,
smear & culture of ascitic fluid, CSF & BM were neg. about acid fast bacilli
Hepatosplenomegaly
lstorage
diseases:
lGaucher
disease
lneonatal
iron storage disease
l Secondary
or metastatic processes
Lymphoma, Leukemia,
Langerhans cell Histiocytosis (bone survey—skin manifestation)
lHemoglobinopathies:thalassemia
major
lCastlemann
‘s disease
lHyperreactive
malaria splenomegaly
lInfections:
lGram
neg. bacteria (Salmonella)
l
Kala Azar (lymphopenia)
lMiliary
TB, Malaria
lCongenital
cytomegalovirus (CMV)
lHepatitis
B, and C
lEpstein-Barr
virus (EBV)
lViral
hemophagocytic syndromes: CMV, EBV, HHV-6
lHuman
immunodeficiency virus (HIV)
lALPS
(Autoimmune lymphoproliferative syndrome)
hemolysis
skin
manifestation
lHLH
(hemophagocyticlymphohistiocytosis)
l
Bicytopenia
lConstitutional
(Inherited)
lVIRUSES
CMV, EBV, Hepatitis
B Hepatitis C
non-B, non-C
(seronegative hepatitis)
HIV
lMARROW
REPLACEMENT
Leukemia , Myelodysplasia, Myelofibrosis
lHLH
HYPERTRIGLYCERIDEMIA
l Hemophagocytic
lymphohistiocytosis (HLH)
l AIDS
lprotease
inhibitions
lAcquired
Immunodeficiency Syndrome
(Human Immunodeficiency Virus)
lVertical
transmission of HIV:
lintrauterine
l
intrapartum
lbreast-feeding
l3
distinct patterns of disease were described in children
l1.
rapid disease course
l2.
slow progression of disease
l3.
long-term survivors
lApproximately
15–25% of HIV-infected newborns in developed countries
present with a rapid disease course,
with onset of AIDS and symptoms during the 1st few months of life and, if
untreated, a median survival time of 6–9 mo
lIn
most infants, physical examination at birth is normal
l
Initial symptoms may be
-subtle,
such as lymphadenopathy andhepatosplenomegaly,
-nonspecific,
such as FTT, chronic or recurrent diarrhea, interstitial pneumonia, or oral
thrush, and may be distinguishable only by their persistence.
Diagnosis
lAll
infants born to HIV-infected mothers test antibody-positive at birth because
of
passive transfer of maternal HIV antibody across the placenta during
gestation
lDefinitive
diagnosis in most infected infants by 1–6 mo of age:
lViral
diagnostic assays, such as HIV DNA or RNA PCR, HIV culture, or HIV p24
antigen immune-dissociated p24 (ICD-p24), are considerably more useful in
young infants
lInfants
born to HIV-infected mothers should be prescribed zidovudine (ZDV)
prophylaxis.
Treatment
lThe
currently available therapy does not eradicate the virus and cure the
patient
lIt
only suppresses the virus for extended periods of time and changes the
course of the disease to a chronic process
(HLH)
Hemophagocytic lymphohistiocytosis
Diagnostic Criteria for HLH:
lFever
>38.5°C and lasting ≥7 days
lSplenomegaly
>3 cm
lTWO
OF THE FOLLOWING HEMATOLOGIC ABNORMALITIES:
Anemia (<9 g/dL hemoglobin)
Thrombocytopenia (<100,000 cells/L)
Neutropenia (<1000 neutrophils/L)
lONE
OF THE FOLLOWING ABNORMALITIES:
Hypertriglyceridemia >2.0 nmol/L
Hypofibrinogenemia <150 mg/dL
land
lHemophagocytosis
in bone marrow, spleen, or lymph node
lNo
evidence of marrow hyperplasia or malignant neoplasia
lHLH
may be present in the absence of genetic mutations of the perforin or Munc
13–4 genes and the presence of
5 of the
following:
lfever,
lsplenomegaly,
lcytopenia
of 2 cell lines,
lhypertriglyceridemia
or hypofibrinogenemia,
lhyperferritinemia,
l
elevated SCD25 (interleukin-2 receptor),
lreduced
or absent NK cells,
land
bone marrow, cerebrospinal fluid or lymph node evidence of hemophagocytosis
Minor criteria
l1.
hyperferritinemia (>500
micgr/dl)
l2.
elevated SCD25 (interleukin-2 receptor) >2400 u/ml
l3.
reduced or absent NK cells
l
@ 2
of these criteria can be substituted for 1 major criteria
lFamilial:
(FHLH)
lSecondary:
Infectious agents:
viruses (e.g., CMV,EBV, parvovirus B19, HHV6), fungi, parasite, protozoa, and
bacteria (Gram neg. ,TB)
HAART
Kawasaki disease
X-linked
lymphoproliferative disorders
Recipients of liver
& kidney Transplantation
EBV associated
limphoproliferativedisorders
lPresentation
with fever, maculopapular and/or petechial rash, weight loss, and
irritability
lFHLH
also is characterized by severe immunodeficiency.
lChildren
with FHLH
always are <4 yr of age, whereas children with secondary HLH
may present at an older age.
lPhysical
examination
:
hepatosplenomegaly,
lymphadenopathy,
respiratory distress,
and symptoms of CNS involvement
laboratory
findings
lAssociated
laboratory findings
in both forms of
HLH include:
hyperlipidemia,
hypofibrinogenemia,
elevated levels of hepatic enzymes,
extremely elevated levels of circulating soluble interleukin-2 receptors
released by the activated lymphocytes,
very high levels of serum ferritin (often >10,000),
and cytopenias (especially pancytopenia from hemophagocytosis in the marrow)
lgenetic
markers for FHLH
can complement a positive family history for other affected children
lTreatment
of the underlying infection, coupled with supportive care, is critical
limmunosuppressive
treatment :
etoposide,
corticosteroids, and intrathecalmethotrexate
lSome
recommend antithymocyte globulin and cyclosporine for maintenance therapy
lEven
with chemotherapy, FHLH remains ultimately fatal, often after a relapse of
the disease.
lAllogeneic
stem cell transplantation is effective in curing
approximately 60% of patients with FHLH
lIn
contrast, in
secondary HLH, when an infection can be
documented and effectively treated, the prognosis is good without any other
specific treatment
lWhen
a treatable infection cannot be documented,
the prognosis may be as poor as that of FHLH, and an identical
chemotherapeutic approach, including etoposide, is recommended