|
پروفسور محمد
حسین سلطان زاده
استاد دانشگاه علوم پزشکی شهید بهشتی
متخصص کودکان ونوزادان
طی دوره بالینی عفونی از میوکلینیک آمریکا
دبیر برگزاری کنفرانس های ماهیانه گروه اطفال
دانشگاه علوم پزشکی شهید بهشتی
|
دکترسید
محمد تقی حسینی طباطبایی
فوق تخصص کلیه اطفال
به اتفاق اعضای
هیئت علمی گروه کودکان
بیمارستان امام حسین
|
تشخیص
¨
Chronic Kidney Disease(CKD)
¨
Is when
one suffer from gradual and usually permanent loss of kidney function over time.
This happens gradually over time, usually month to years.
¨
KIDNEY FAILURE
¨
Occurs
when the kidney partly or completely lose their ability to carry out normal
function.
¨
DEFINITION: GFR < 15 ml / min / 1.73m2, with sign and symptoms of uremia or the
need for dialysis / transplant for treatment of complications of decreased GFR
that increase risks of mortality and morbidity.
¨
ESRF, is
administrative term in the USA, for the payment of healthcare by the Medicare
ESRF program.
¨
1 – RENAL
IMPAIRMENT. GFR = > 80ml, but loss of reserve function.
¨
2 – MILD
RENAL INSUFFICIENCY. GFR = 50 – 80ml, renal mass is 25 – 50%.
¨
3 –
MODERATE RENAL INSUFFICIENCY. GFR = 30 – 50ml, renal mass is 15 – 25%.
¨
4 – SEVER
RENAL INSUFFICIENCY, GFR = 10 – 30ml, renal mass is 5 – 15%.
¨
5 –
END-STAGE RENAL FAILURE. GFR < 10ml, renal mass<5%.
¨
CKD:
¨
Stages of
chronic kidney disease:
1 –
kidney damage with normal GFR, ( > 90 ml ).
2 –
kidney damage with mildly decrease GFR, as
( 60 – 89 ml ).
3 –
moderate decrease in GFR, as ( 30 – 50 ml ).
4 –
sever decrease in GFR, ( 15 – 29 ml ).
5 –
kidney failure, GFR < 15 ml or on dialysis.
It is
possible to have a GFR 60 -89ml/min/1.73m2 without kidney damage. In INFANT,
VEGETRIAN DIET, UNILATERAL NEPHRECTOMY, VOLUME DEPLETION, HEART FAILURE.
¨
Cause of CKD:
¨
1 –
Congenital abnormalities.
¨
2 –
Hereditary condition.
¨
3 –
Glomerulonephritis.
¨
4 –
Multisystem disease.
¨
5 –
Miscellaneous:
-renal vascular disease,
-kidney tumor,
-drash syndrome,
-others.

¨
Case
presentation:
¨
a 13
years-old girl presented with increasing fatigue, anorexia, weight loss, and
sever pallority. She also had a history of polyuria, polydipsia, since infancy,
and enuresis, nocturia. Her family history was unremarkable.
¨
Serum
Creatinine was markedly elevated, and urinalysis showed a low SG, with mild
proteinuria and glycosuria. US showed small echogenic kidneys with multiple
small cortico- medullary cysts.
¨
DIFFERENTIAL DIAGNOSIS
¨
1 – CYSTIC
DISEASE: ARPCK.
ADPCK.
GCKD.
SIMPLE CYSTS.
DIFFUSE CYSTIC DYSPLASIA.
CONGENITAL SYNDROMES.
NPHP/MCKD COMPLEX.
CHRONIC PYELONEPHRITIS.
UT OBSTRUCTION.
OLIGOMEGANEPHRONIC
DYSPLASIA.
MEDULLARY SPONGE
KIDNEY
¨
Clinical presentation and evaluation of cystic disease
¨
Renal
cystic disorders represent a heterogeneous group of diseases which may present
in utero or be clinically silent well into adulthood.
¨
The age of
presentation, family history, and other clinical signs and symptoms are
essential in delineating specific disease entities.
¨
Cystic
disorders include: ARPCK, ADPCK, GCKD, diffuse cystic dysplasia, simple cyst,
MCD/JNPH, and ACKD(acquired).
¨
Cystic
kidneys are an important component of several congenital syndrome.
¨
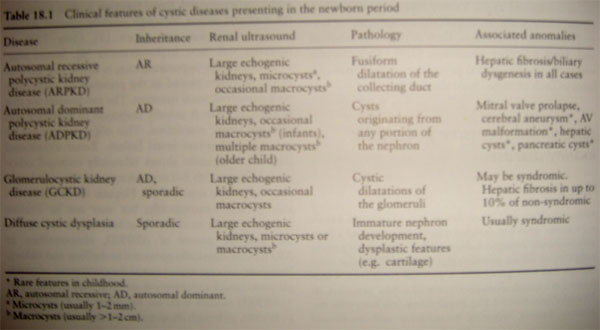
ARPCK: autosomal recessive PCK.
¨
This
presentation of a neonate with oligohydramnios, respiratory distress, large
echogenic kidneys, and HTN. Renal function is normal in in the neonatal period.
¨
Hepatic
fibrosis is invariably present histologically, (abnormality are classically
restricted to the liver and kidney, and occasionally multiple intracranial
aneurysms in the adults).
¨
Detailed
family history, US of the parents, and the presence of associated anomalies are
more helpful in establishing the diagnosis
¨
ADPCK: autosomal dominant PCK.
¨
The
presentation is from neonate to adulthood.
¨
Symptoms
include hematuria, HTN, flank pain, UTI and sterile pyuria.
¨
US
demonstrates one or more macroscopic cysts.
¨
ADPCK is a
systemic disease, with multiorgan system involvement. Intracranial aneurysms,
mitral valve prolapse, liver and pancreatic cyst, ovarian cysts, intestinal
diverticuli, and hernias may also develop.
¨
ADPCK is
responsible for 8 – 10% of cases of ESRF in Europe.
¨
CONT:
¨
Glomerulocystic Kidney Disease: typically present in the neonatal period, with
large echogenic kidneys and microscopic glomerular cysts.
¨
SIMPLE
CYSTS: is age dependent, and occurring < 1% in children.
¨
DIFFUSE
CYSTIC DYSPLASIA: occurs primarily as a sporadic condition, or as a part of
multiple malformation syndromes. In the cysts is primitive elements such as
cartilage.
¨
Acquired
Cystic Kidney Disease: occurs in ESRF, that are on dialysis. And kidney is
enlarged.

¨
NPHP/MCKD complex:
¨
as a
distinct clinicopathologic entity of inherited diseases that lead to CRF, due
to chronic sclerosing TINephropathy. ( cystic formation, tubular atrophy,
tubular basement membrane disintegration, interstitial fibrosis).
¨
NPHP is an
aut. rec. disease with onset of ESRF in adolescence, and is the most frequent
genetic cause of CRF in first two decades of life ( 10 – 20% of all cases ).
¨
MCKD is
used for aut. dom. Variants with onset in adulthood.
¨
NPHP/MCKD,
divided as INFANTILE, JUVENILE, and ADOLESCENT, forms.
¨
INFANTILE
NPHP: NPHP2, in which ESRF, occurs within the first 3 years of life. It has
enlarged kidneys and cortical micro cysts, absence of medullary cysts. Gene
locus was localized to 9q22-q31. (is same as ARPCK).
¨
JUVENILE
NPHP: NPHP1, a gene locus has been mapped to chromosome 2q12.3 (NPHP1). Some of
patients has extra-renal manifestation.
¨
ADOLESCENT
NPHP: NPHP3, has been localized to chromosome 3q21-q22.
¨
The
disease genes for NPHP4 – 8 has been identified.
¨
The
proteins for which they encoded are known, as the NEPHROCYSTINS, that localized
at least in part to an organelle in the cell called the primary CILIA.
¨
NPHP/MCKD complex: clinical
manifestation,
¨
This
diseases characterized by the insidious onset of renal failure.
¨
In
recessive NPHP, symptoms of polyuria, polydipsia, decrease urinary concentrating
ability, 2nd enuresis, are the earliest presenting symptoms in more
than 80% of cases, and occur at 4 – 6 years of age.
¨
Starting
to regularly drink at night at 6 – 10 years old.
¨
Pallor,
weakness, and pruritus are common.
¨
Anemia and
growth retardation occur latter and are usually pronounced.
¨
Median age
for ESRF is 13 years.
¨
Typically,
edema, hematuria, and UTI are absent in NPHP.
¨
HTN is
rare ( for degree of CRF ).
¨
Most
children have already develop CRF when the first come to clinical attention.
¨
They have
definite risk of sudden death from fluid and electrolyte imbalance (rare).
¨
If RF has
not developed by the age 25, the diagnosis of rec. NPHP should be questioned.
¨
Disease
recurrence has never been reported after renal transplantation.
¨
EXTRA-RENAL manifestation, occur only in recessive NPHP, but is absent from MCKD.
1 – retinitis pigmentosa.
2 – mental retardation.
3 – skeletal changes.
4 – cerebellar ataxia.
5 – liver fibrosis.
6 – retinal degeneration.
7 – polydactyly.
¨
DIAGNOSIS:
¨
as
increase serum Creatinine value in patients with nonspecific complaints.
¨
Prolonged
nocturia since school age.
¨
Low early
morning urine SG.
¨
Small to
normal size and echogenic kidneys.
¨
Confirmed
by molecular genetic diagnosis in NPHP.
¨
Ophthalmoscopy should be performed.
¨
Liver
function tests and hepatic US, are important.
¨
Kidney US,
or MRI. For detection of renal cysts.
¨
Kidney
biopsy is not initial procedure (due to PCR).
¨
PROGNOSIS and THERAPY:
¨
There is
no specific therapy.
¨
Therapy is
symptomatic.
¨
Psychological counseling.
¨
Renal
replacement therapy.
¨
All
patients develop ESRF in first 2 decades.