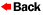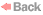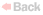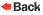|
پروفسور محمد
حسین سلطان زاده
استاد دانشگاه علوم پزشکی شهید بهشتی
متخصص کودکان ونوزادان
طی دوره بالینی عفونی از میوکلینیک آمریکا
دبیر برگزاری کنفرانس های ماهیانه گروه اطفال
دانشگاه علوم پزشکی شهید بهشتی
|
معرفی : دکتر
شاداب صالح پور
فوق تخصص غدد اطفال
به اتفاق اعضای هیئت علمی گروه کودکان
بیمارستان مفید
|
تشخیص
بیماری گوشه
Gaucher Disease
Answer:
This nine-year-old girl, who had a long history of
epistaxis and hepatosplenomegaly, presented with acute leg pain.
Interestingly, a year earlier she had had similar leg pain, but it
had resolved without intervention. An important observation is that
the hepatosplenomegaly was chronic and probably progressive.
Pertinent features of the history include the absence of fevers,
trauma, scleral icterus, jaundice, weight loss, visual disturbances,
medications, and previous health problems. The results of the
physical examination are notable for the presence of low-grade fever,
mild systolic hypertension, marked hepatosplenomegaly, and tenderness
in the right thigh. The results of the neurologic examination were
normal, with no ocular motor abnormalities. Laboratory screening
revealed slight anemia, leukocytosis, low iron saturation, and a high
erythrocyte sedimentation rate, with minor elevations in the levels
of lactate dehydrogenase and aminotransferases. Cultures of blood and
urine were negative.
Abdominal ultrasonographic studies confirm the
presence of hepatosplenomegaly. A pelvic CT scan showed an effusion
in the right hip and patchy sclerosis in the left femoral epiphysis,
which suggested a previous episode of avascular necrosis.
MRI of the
pelvis and femurs was performed before and after the administration
of gadolinium. On T1-weighted images, the normal fatty
marrow of the pelvis and femurs is diffusely replaced by abnormal
marrow. The fatty marrow should have the same signal intensity as
subcutaneous fat, which appears as a bright stripe on this image.
Instead, it has the same signal intensity as muscle, indicating that
the bone marrow is affected by an infiltrative process. On a T2-weighted
image, there is edema in the marrow of the proximal right femur and
in the surrounding soft tissues; the scan obtained after the
administration of gadolinium shows diminished enhancement of this
region. These findings are characteristic of an acute bone infarct.
The distal femoral metaphyses are flared, an effect that is probably
due to the diffuse marrow infiltrate.
The two
central features of this case are chronic hepatosplenomegaly and
acute bone pain, which appears to be intermittent in nature. The
imaging studies suggest the presence of an infiltrative process in
the liver and the bone. I will consider the broad differential
diagnosis of hepatosplenomegaly and focus on disorders that can also
account for the bone findings, assuming that the hepatosplenomegaly
and the bone pain are the result of a single disease process. The
causes of hepatosplenomegaly can be sorted into broad categories that
include anatomical abnormalities, congestion, infection, hematologic disorders,
and infiltrative processes.
Several
specific anatomical entities, such as cysts, malformations, and
developmental anomalies, may result in hepatomegaly, splenomegaly, or
bone pain. However, these entities are usually regional and are not
likely to result in the constellation of findings that are present in
this case. Furthermore, most anatomical anomalies have characteristic
features on imaging studies, and such features are not apparent on
this patient's radiographic studies.
Passive
venous congestion can certainly cause long-standing
hepatosplenomegaly, which can be progressive. Congestion cannot
account for this child's bone pain, however, and furthermore there
are no additional signs of congestion, such as venous engorgement,
edema, stasis of the legs, or evidence of esophageal varices.
Infection
seems unlikely in this case for three reasons. First, although
several infections are known to cause either hepatosplenomegaly or
bone pain, it would be distinctly unusual for infection to cause both
of these clinical findings. Second, many infections tend to occur
over a relatively short period, whereas this child's organomegaly was
chronic. Third, most infections are associated with increased blood
flow, which would typically cause increased uptake rather than
decreased uptake on bone scanning.
Chronic
hepatosplenomegaly is a feature of several hematologic diseases,
including chronic hemolytic anemia, disorders associated with
extramedullary hematopoiesis, and myeloproliferative disorders.
Chronic hemolytic anemia usually causes hyperbilirubinemia, anemia,
and reticulocytosis, none of which were present in this case. In
addition, splenomegaly is usually more prominent than hepatomegaly in
chronic hemolytic anemia.
Sickle cell
disease deserves consideration, since this child's acute bone pain is
suggestive of a vaso-occlusive crisis. However, her medical history
includes no other manifestations of sickle cell disease, and this
disorder would not account for the progressive organomegaly.
Splenomegaly is a common finding in early childhood, but as children
approach their teenage years, it is very uncommon. Although
intrahepatic sickling may lead to some degree of liver enlargement,
it usually develops rapidly and typically is associated with evidence
of liver dysfunction.
Disorders
associated with extramedullary hematopoiesis, such as osteopetrosis
and myelofibrosis, may be associated with chronic hepatosplenomegaly,
but in patients with such disorders one would expect characteristic
findings in the peripheral blood and on the radiographs. Finally,
myeloproliferative disorders often lead to organomegaly, but they too
are associated with additional findings, such as marked abnormalities
in the peripheral-blood counts.
Infiltrative processes that cause hepatosplenomegaly and bone pain
include both malignant and nonmalignant conditions. Childhood cancers
associated with hepatosplenomegaly and bone pain include leukemia,
lymphoma, neuroblastoma, and primary hepatic tumors. Cancer seems
highly unlikely in this case simply because of the time course of the
patient's symptoms. Neoplasia in children usually takes the form of a
relatively acute process, with escalating symptoms over the course of
weeks or months rather than years. Despite this general observation,
it is important to consider briefly the specific malignant conditions
that could be relevant to this case.
Hepatosplenomegaly and bone pain are common findings in children with
leukemia; however, there are usually additional findings, such as
fever, an appearance of illness, and cytopenia.1
Lymphomas in children may cause hepatosplenomegaly, but they have
additional clinical features as well, such as fever, chills, weight
loss, and marked lymphadenopathy. Hepatosplenic T-cell lymphoma is
a unique form of non-Hodgkin's lymphoma that may cause marked
hepatosplenomegaly without lymphadenopathy. However, this disease
typically affects young men, who present with prominent fever, weight
loss, and cytopenia.
Metastatic
neuroblastoma often involves both the liver and bones. This degree of
hepatomegaly in neuroblastoma is most commonly associated with stage
IV-S disease, which occurs exclusively in infants. In older children,
stage IV neuroblastoma may include both bone pain and hepatomegaly;
however, these children appear very ill, the bone involvement is
typically multifocal, and there is avid uptake of labeled technetium
on bone scanning. Hepatoblastoma may cause liver enlargement and may
metastasize to the bones, but this disease occurs almost exclusively
in children less than two years of age, and the vertebrae are the
usual metastatic targets.
Histiocytic
disorders that can occur in children include Langerhans'-cell
histiocytosis, non-neoplastic histiocytic disorders, including
familial hemophagocytic lymphohistiocytosis and the infection-associated
hemophagocytic syndrome, and metabolic storage diseases. Langerhans'-cell
histiocytosis is a neoplasm of the Langerhans' cells that has
protean clinical manifestations. Common clinical findings include
lytic bone lesions, dermatitis, organomegaly, lymphadenopathy, fever,
weight loss, chronic otitis media, and diabetes insipidus.
Disseminated disease is seen in young children but is unusual at this
patient's age. The absence of these typical clinical findings, along
with evidence of a bone infarct, makes Langerhans'-cell histiocytosis
very unlikely. Patients with familial hemophagocytic
lymphohistiocytosis may present with infiltrative hepatosplenomegaly,
but most often they present (earlier in life than the child in this
case) with rapid clinical deterioration, including fever, wasting,
and skin rash, irritability, and central nervous system findings.
Likewise, the infection-associated hemophagocytic syndrome usually
follows a viral process, involves fever and pancytopenia, and usually
occurs in the setting of immunodeficiency, which does not seem to be
present in this case.
Hepatosplenomegaly is a common feature in many metabolic storage
diseases, ranging from sphingolipidosis to mucopolysaccharidosis. A
smaller number of metabolic disorders are associated with bone pain
or, as it is often called, bone crisis. The combination of
hepatosplenomegaly and bone pain is unique and is a feature of
Gaucher's disease. Gaucher's disease is a lysosomal glycolipid-storage
disease characterized by the accumulation of glucocerebroside
in various tissues as a result of a deficiency in the enzyme beta-glucosidase.
There are three forms of Gaucher's disease. The most common form,
non-neuronopathic (or type I) disease, involves the progressive
engorgement of macrophages over time, a process that results in
organomegaly, bone marrow infiltration, and damage to the bones,
which may lead to infarction or fracture. Children with this form of
Gaucher's disease are healthy in the first few years of life;
hepatosplenomegaly then gradually develops over the course of several
years. Splenomegaly is usually more prominent than hepatomegaly, but
there is notable heterogeneity in the clinical manifestations of
Gaucher's disease.
Bone involvement is present in nearly all patients and typically
occurs after visceral disease. The classic Erlenmeyer-flask
deformity, a common radiographic finding, is caused by medullary
expansion and remodeling of the distal femur due to the accumulation
of histiocytes. Patients usually have episodic, painful bone crises,
which have a predilection for the femoral head and sometimes are
misdiagnosed as Legg–Calvé–Perthes disease.
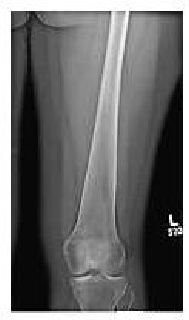
Erlenmeyer flask deformity of
the distal
These bone
crises result from ischemia and infarction due to progressive marrow
infiltration with enlarging storage cells. This patient's current
episode of leg pain, as well as the episode one year earlier,
represents a bone crisis that is fairly typical of Gaucher's disease.
Classic features of bone crisis in Gaucher's disease include fever,
leukocytosis, an increased erythrocyte sedimentation rate, and
absence of tracer uptake on bone scans (i.e., "cold" bone scans).
Patients with Gaucher's disease are at increased risk for malignant
hematologic tumors that may cause bone pain, but such tumors tend to
occur much later in life and typically show increased tracer uptake
on bone scans.
In addition
to hepatosplenomegaly and bone pain, this patient had a three-year
history of epistaxis. Bleeding, particularly epistaxis, is common in
patients with Gaucher's disease and is usually the result of
thrombocytopenia due to hypersplenism. Qualitative defects in
platelet function and decreased levels of several coagulation factors
have also been described in patients with this disease.
This
patient probably has type I, or non-neuronopathic, Gaucher's disease,
since the rarer form — type II, or neuronopathic, Gaucher's disease —
is severe, involves the central nervous system, and occurs in young
children, who usually die by the age of two years. Type III, or
subacute neuronopathic, Gaucher's disease cannot be definitively
ruled out because neurologic signs may develop after symptomatic bone
disease. Ocular motor manifestations, which are typically the first
evidence of neurologic involvement, were not present in this case.
Gaucher's
disease can be diagnosed on the basis of a low level of beta-glucosidase
in peripheral-blood leukocytes. An alternative and more expeditious
diagnostic approach is bone marrow aspiration to search for classic
Gaucher's cells, which are very large macrophages with characteristic
morphologic features. With regard to clinical management, bony crises
typically resolve over the course of a few days with adequate
hydration and analgesia. Corticosteroids may also be beneficial.
Overall, this patient has an excellent prognosis and should be a
candidate for long-term enzyme replacement, which effectively
reverses the manifestations of Gaucher's disease and prevents
long-term complications.
Clinical Diagnosis: Gaucher's
disease.
The
diagnostic procedure was bilateral bone marrow biopsy and aspiration
of the iliac crest. The tissue specimens retrieved from the two sites
had similar histologic features. The marrow contained solid sheets or
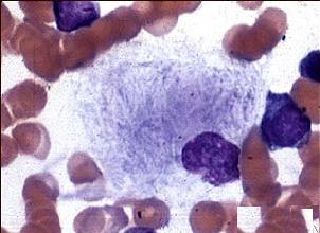
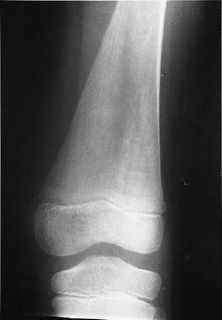
clusters of large cells or single large cells that displaced most of
the hematopoietic elements. These cells, with a diameter that was
about two to six times that of normal macrophages, contained
round or oval vesicular nuclei and abundant eosinophilic cytoplasm
with a fibrillar or striated appearance. Wright–Giemsa staining of a
smear of aspirate also showed numerous large cells with striated
cytoplasm. These cells are histiocytes, and their large size and
fibrillar cytoplasm are characteristic of Gaucher's cells. The
differential diagnosis includes other metabolic storage diseases and
secondary accumulation of Gaucher's cells. The other major storage
disease to consider is Niemann–Pick disease. In that disorder, the
large histiocytes that accumulate have foamy cytoplasm with round
vacuoles, rather than a fibrillar or striated appearance. Gaucher's
cells — or pseudo-Gaucher's cells, as they are sometimes called — may
be found in persons without beta-glucosidase deficiency when
there is a very high rate of cell turnover. The classic setting for
this is chronic myelogenous leukemia, in which the presence of
increased numbers of hematopoietic-cell precursors in the marrow can
overwhelm the normal cellular machinery and result in the
accumulation of Gaucher's cells.
The
combination of the clinical findings and the morphologic features of
the bone marrow specimens from the iliac crest confirm that this
patient's diagnosis is type I Gaucher's disease. Gaucher's disease is
the most common of the genetic lysosomal storage diseases, affecting
1 in 50,000 to 100,000 live births; it can be inherited in an
autosomal recessive fashion or acquired through a spontaneous
mutation. Glucocerebroside, a normal component of the cell membrane,
is generated in macrophages as they degrade effete cells during
routine cell turnover. In the absence of beta-glucosidase,
glucocerebroside accumulates in the lysosomes of macrophages, where
it is stored in a specific architectural arrangement, resulting in
elongation of the lysosomes. These elongated and distended lysosomes
fill the cytoplasm of the macrophages, giving it the characteristic
fibrillar or striated appearance, which has been likened to wrinkled
cigarette paper.
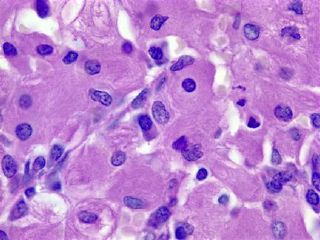
Gaucher's cells